
Science Behind Tangent Bioworks
On-Demand Molecular Dynamics Simulations
Run reliable protein molecular dynamics simulations using GROMACS without maintaining your own HPC infrastructure.
We provide a secure, researcher-focused platform to perform production-grade molecular dynamics (MD) simulationsfor proteins using GROMACS/DESMOND, charged transparently by simulation time.
What Tangent Offer
Protein Molecular Dynamics as a Service
We run end-to-end MD simulations on high-performance compute systems and deliver complete, ready-to-analyze results.
Our service includes
Protein MD simulations using GROMACS
Standard, validated MD workflows
Secure handling of uploaded structures
Downloadable trajectories and analysis outputs
Clear job tracking and queue transparency
You upload your structure.
We handle the computation.
Why use Tangent platform
No infrastructure overhead, we run everything for you
Scientifically standard workflows (GROMACS-based)
Pay only for what you use
Queue transparency and job tracking
Research-only, confidentiality-focused service
End-to-End Simulation Pipeline
Tangent validated, reproducible molecular dynamics workflow based on best practices in the MD community
find the flow
click here
click here
Quick Molecular Dynamics Simulation Submission
│
▼
Input Validation
(PDB / CIF / SDF / MOL2)
│
▼
Structure Preparation
│
├─ Protein Preparation
├─ Ligand Preparation
└─ Mutation Modeling (Optional)
│
▼
Complex Construction
│
▼
Force Field Assignment
│
▼
System Building
│
├─ Solvation
├─ Ion Addition
└─ Box Generation
│
▼
Energy Minimization
│
▼
NVT Equilibration
│
▼
NPT Equilibration
│
▼
Production MD Simulation
│
▼
Trajectory Processing
│
▼
Structural Analysis
│
├─ RMSD
├─ RMSF
├─ Rg
└─ SASA
│
▼
Interaction Analysis
│
├─ Hydrogen Bonds
├─ Salt Bridges
├─ Contact Mapping
└─ Interaction Networks
│
▼
Conformational Analysis
│
├─ PCA
├─ Clustering
├─ DCCM
└─ Free Energy Landscape
│
▼
Binding Energy Analysis
│
├─ MM/PBSA
└─ MM/GBSA
│
▼
Result Compilation
│
▼
Output Generation
│
├─ Scientific Report (PDF)
├─ Publication Figures
├─ Analysis Data
├─ Simulation Trajectories
└─ Structure Files
│
▼
Final Results
Detailed Project Submission
│
▼
Input Validation
(PDB / CIF / SDF / MOL2)
│
▼
Research Information Collection
│
├── Research Area
├── Scientific Question
├── Expected Outcome
└── Project Description
│
▼
Molecular System Identification
│
├── Protein Only
├── Protein-Ligand Complex
├── Protein-Protein Complex
├── Membrane Protein
├── Antibody-Antigen Complex
├── Peptide System
├── Nucleic Acid System
└── Custom System
│
▼
Structure Availability Check
│
├── PDB ID Available
├── Structure File Uploaded
└── Custom Structure Provided
│
▼
System Components Extraction
│
├── Protein Name
├── Gene Name
├── Organism
├── Ligand Information
├── Mutations
└── Membrane Composition
│
▼
Simulation Configuration
│
├── Simulation Length
├── Force Field Selection
└── Special Requirements
│
▼
Analysis Selection
│
├── RMSD
├── RMSF
├── Radius of Gyration
├── Hydrogen Bond Analysis
├── PCA
├── MM/PBSA
├── Contact Analysis
├── Interaction Network
├── Clustering
└── Trajectory Visualization
│
▼
File Collection
│
├── Protein Structure
├── Ligand Structure
├── Input Files
├── Supporting Documents
└── Published References
│
▼
Project Classification
│
├── Standard MD
├── Mutation Study
├── Binding Study
├── Membrane Dynamics
├── Drug Discovery
└── Custom Workflow
│
▼
Workflow Recommendation
│
▼
Simulation Pipeline Generation
│
▼
Analysis Pipeline Generation
│
▼
Scientific Output Package
│
├── Report
├── Figures
├── Trajectory Files
├── Structure Files
├── Analysis Data
└── Publication Materials
│
▼
Final Results
or
Already have a protein, ligand, or complex structure?
Skip the form and email your files directly to:
commercials
1₹(0.0106$) per 100 ATOMS per 1 NANOSECOND
<25,000 atoms
₹500
10 ns
₹1,000
25 ns
₹1,500
50 ns
₹2,500
100 ns
Custom Quote
>100 ns
25,000–50,000
₹1,000
10 ns
₹1,500
25 ns
₹2,500
50 ns
₹3,500
100 ns
Custom Quote
>100 ns
50,000–100,000
₹1,500
10 ns
₹2,500
25 ns
₹3,500
50 ns
₹5,000
100 ns
Custom Quote
>100 ns
>100,000
Custom Quote
10 ns
Custom Quote
25 ns
Custom Quote
50 ns
Custom Quote
100 ns
Custom Quote
>100 ns
Custom Quote Request
Need a simulation beyond our standard pipeline?
Submit your project details, molecular structures, or research objectives, and our team will prepare a customized quotation based on: Molecular system complexity - Number of atoms - Simulation duration - Membrane requirements - Enhanced sampling methods - Free-energy calculations - Custom analyses and deliverables - Computational resource requirements
Every project is unique. We will review your submission and provide a customized simulation strategy, timeline, and cost estimate.
Request a Custom Quote
Upload your files or email your project details to:
sample output
Protein - Small molecule simulation report
plot based













Protein - DNA molecule simulation report
plot based

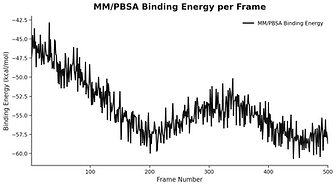
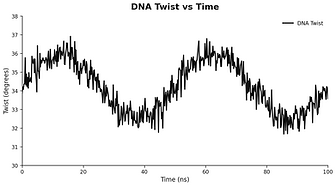






















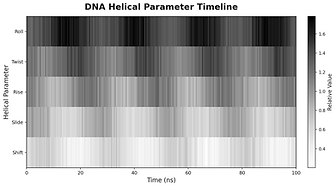
Your Questions, Our Science
How long does molecular docking take?
Most molecular docking projects are completed within 24–72 hours, depending on the number of compounds, target complexity, and required analyses. Large-scale virtual screening projects may require additional time.
What files do I need to provide?
To begin a project, we typically require:
-
Protein structure files (PDB format) or target information
-
Ligand structures (SDF, MOL2, PDB, or SMILES format)
-
Project objectives and specific requirements
If you do not have these files, our team can assist with data preparation.
Do you sign Non-Disclosure Agreements (NDAs)?
Yes. We understand the importance of research confidentiality and are happy to sign NDAs before discussing sensitive academic, industrial, or proprietary projects.
Can you support scientific publications?
Yes. We provide publication-ready figures, statistical analyses, methodology descriptions, and result interpretation suitable for manuscripts, theses, dissertations, and grant applications.
Do you provide raw data and project files?
Yes. Upon project completion, clients receive all relevant raw data, processed results, simulation trajectories, structural models, figures, and technical reports generated during the study.
What deliverables will I receive?
Deliverables may include:
-
Detailed technical reports
-
Publication-ready figures
-
Docking poses and interaction analyses
-
Molecular dynamics trajectories
-
Binding energy calculations
-
Processed datasets and raw output files
Can you customize analyses for my research project?
Absolutely. Every project can be tailored to your research objectives, whether it involves drug discovery, structural biology, bioinformatics, cancer research, immunology, or multi-omics data analysis.
Do you offer support after project delivery?
Yes. We provide post-delivery support to answer questions, clarify results, and assist with interpretation of findings.

Powered by High-Performance Cloud Computing
Our computational workflows are executed on high-performance cloud infrastructure utilizing compute-optimized, memory-optimized, and GPU-accelerated instances, including configurations comparable to AWS C-series, M-series, R-series, and NVIDIA GPU-enabled environments. This enables efficient processing of large molecular simulations, bioinformatics pipelines, and multi-omics datasets without requiring researchers to maintain their own HPC infrastructure.